Stephane Wenric, PhD
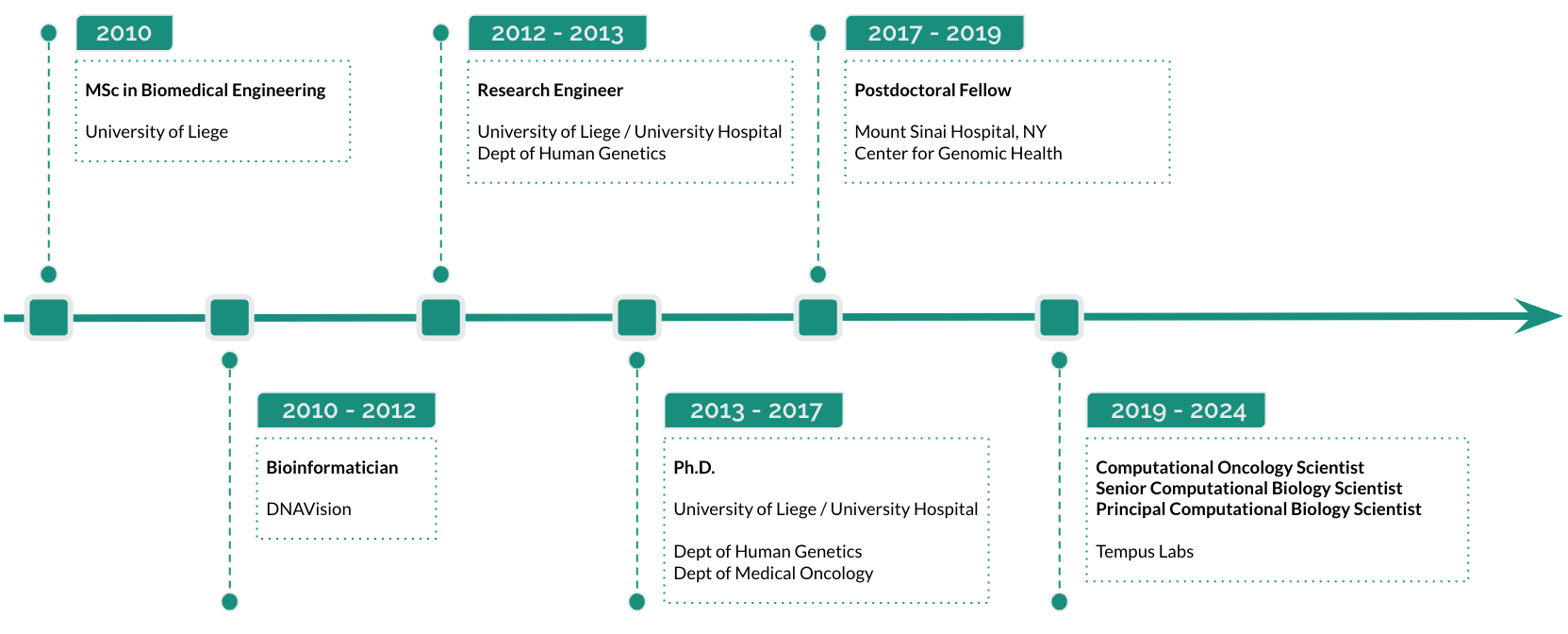
Experienced computational biologist and team lead with a strong scientific and technical background including 12 years of expertise in genomics/transcriptomics, clinical data and oncology, machine learning and statistics, and client-facing experience.
I currently work as a Principal Scientist - Computational Systems Biology at Tempus.
Previously, I was a Postdoctoral Fellow at the Icahn School of Medicine at Mount Sinai, NY.
I hold a PhD in Biomedical Sciences (bioinformatics, cancer genomics) and a master's degree in Biomedical Engineering from the University of Liège
I have authored 15+ publications in peer-reviewed scientific journals, one US patent, one European patent, I have presented scientific results at several international conferences, and I have received more than $100K in research grants and academic excellence awards, including a BAEF Fellowship.